advertisement



Clinician Scientist Lecture: The Immune System in Glaucoma
AGS 2003 Meeting - March 2003, San Francisco, USA
Martin B. Wax
In the 2003 Clinician-Scientist lecture at the AGS, I provided an overview of the roles of the immune system in glaucoma. Several lines of evidence suggest that one prominent role of the immune system is one of surveillance where it acts as a key modulator in retinal ganglion cell fate decisions in response to stress, Another key role is to act as a modulator of both autoprotective and autoadverse immunity. It is generally accepted that the main retinal/optic nerve stress conditions that occur in glaucoma are elevated intraocular pressure (IOP) and ischemia. Both of these were described over 150 years ago. In the past decade, good evidence has accumulated to support a role for additional stressors that may include oxidative stress and free radical formation, excitotoxicity (still quite controversial), inflammatory cytokines (TNF-α and nitric oxide), and aberrant immunity.
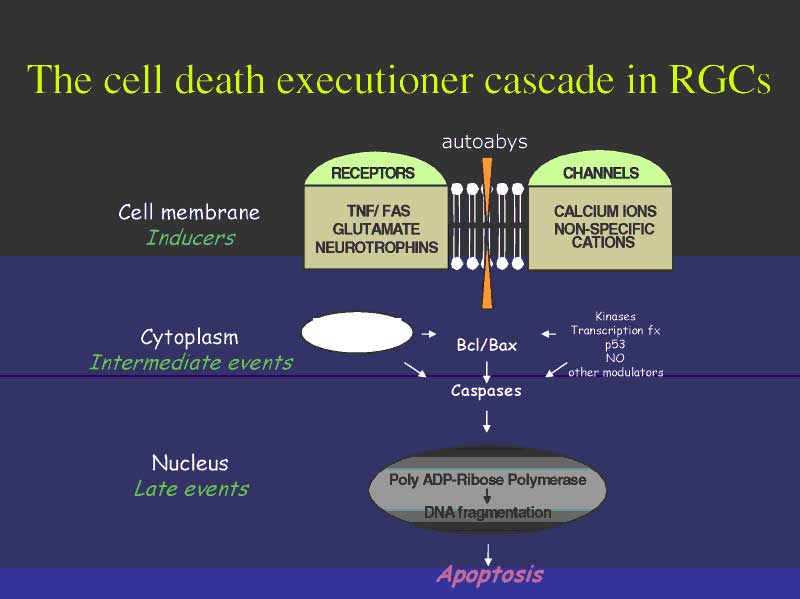
Bill Tatton has beautifully diagrammed many of the presumed stress conditions in glaucoma coupled to immune pathway signaling cascades in Figure 1. If we consider that a fundamental 'thermostat' that regulates cell fate decisions is the relative ratio of the two cytoplasmic proteins Bcl and Bax (or their isoforms), then an elegant simplicity found in Bill s diagram readily emerges. Although the apoptosis cascade is intricately regulated by both stimulatory and inhibitory pathways, there are basically two ways to intervene to alter the apoptosis cascade. One can envision designing neuroprotective strategies to treat glaucoma using therapies that either bolster the endogenous neuroprotectant Bcl, by intervening in the lower half of the diagram, or by intervening to suppress the noxious substrate Bax in the top half of the diagram. Interventions that are designed to increase Bcl components appear, in general, to utilize intracellular manipulations that involve gene therapy, whereas those that attempt to minimize Bax production may be small molecule based, and targeted to specific cytokine cascades. In either case, it should be readily apparent that indeed the immune system is critical to determining RGC survival in homeostasis and stress conditions, thus supporting the argument that the immune system serves a key surveillance role that regulates cell fate under conditions commonly found in the glaucomatous eye.
The role of the role of protective autoimmunity in modulating RGC cell fate has recently been well reviewed by Michal Schwartz in numerous publications. Regarding autoimmunity, there are several reasons we suspect that some forms of glaucoma, particularly in patients whose IOP is normal, represents an autoimmune neuropathy. The evidence supporting the existence of the entity we term 'presumed autoimmune glaucoma', is derived from a substantial body of clinical and bench work over the past eightyears. Some of this evidence includes the following findings:
- There is an epidemiological association of immune-related disease
in patients with NPG.
In a study by Anderson et al., researchers found that 30% of patients with NPG also have other autoimmune diseases whereas, in the control population, only 8% of individuals had other autoimmune diseases. However, this does not mean that if a person has NPG, he will definitely contract another autoimmune disease or vice versa, though it is certainly true that you can have more than one autoimmune illness. Indeed, autoimmune disease can be associated with a host of other systemic diseases, but it can also be very organ-specific. Therefore, the only manifestation of autoimmune disease in some patients might be NPG. This is not to say that the presumed autoimmune form of glaucoma only occurs in patients with NPG, although we do believe that it is more prevalent in that subgroup. Patients with high-IOP glaucoma can essentially have two things wrong: high IOP and an autoimmune disorder. In these patients, it is more difficult to zero in on the autoimmune signal that is buried in the noise of their high pressure. - There are increased aberrant serum autoantibodies, including
monoclonal paraproteins and non-organ-specific antibodies to DNA, RNA
and nuclear proteins, in patients with NPG.
Many patients with normal-pressure glaucoma test positive for autoantibodies that are typically sought for when looking for other autoimmune diseases. (Examples include antinuclear antibodies or autoantigens Ro [SS-A] and LA [SS-B] which are indicative of Sjogren's disease, and may reflect autoantibodies to heat shock protein 60). But the most significant finding in these patients would be the presence of a monoclonal gammopathy. T cells and B cells function to mediate the immune response. B cells function mainly to produce antibodies. In some patients, the B cells go on the rampage, producing inordinate quantities of antibodies. These 'monoclonal antibodies' are a clone of the B cells that went awry. Monoclonal gammopathy is not uncommon, appearing in approximately 2% of elderly Americans. But this figure is much higher, 10-12%, in our normal-pressure glaucoma population. The significance of monoclonal gammopathy is that, in approximately one-third of patients who have them, it may signal an underlying lympoproliferative disorder (i.e., cancer) which, if detected early, can be treated successfully, such as in patients with multiple myeloma. Immunological and neurological research from the mid-1980s suggested that monoclonal antibodies are likely causes of neuropathy. Their insidious destruction of neurons parallels the natural history of glaucoma, a slow and insidious disease. This is certainly an attractive hypothesis, but it is not proof.
Autoimmune diseases are all basically defined by their association in patients who produce relevant antibodies. This increase in antibodies is often good enough for people to label a disease as being 'autoimmune'. But the true gold standard needed to define an autoimmune disease is: can you give the antibody and cause the disease? Or, can you remove the antibody and cure the disease? We believe we now have the data to suggest that giving certain antibodies can elicit an experimental autoimmune glaucoma, at least in rats. In humans, we have tried preventing the production of the antibodies, but these experiments comprised too few patients to really produce a meaningful answer. The problem with glaucoma is that the deterioration is a sensory neuropathy. By removing antibodies, we can do nothing more than stabilize the patient's vision. However, if we were instead dealing with a motor neuropathy, eliminating the appropriate antibodies could enable patients to use their arms again, or to walk again. Trying to establish proof by removing the culprit antibodies in a sensory neuropathy is a therefore a tough hurdle to climb. What is necessary is a very broad study in a large number of patients. Unfortunately, this requires considerable financial support. Since presumed autoimmune glaucoma probably occurs in a small number of glaucoma patients (<5%), it is unlikely that a high funding priority will be obtained from the NIH if such a study were to be proposed. - Elevated serum levels of autoantibodies to 60-kD bacterial and
human heat-shock protein are found in patients with glaucoma.
When our body fights a bacterial infection, our immune system attacks the bacteria and kills and eliminates it from our body. But what is it that our immune system recognizes on the surface of the bacteria that enables this to happen? The largest antigenic protein (recognition site) on the surface of a bacterium is a protein known as heat shock protein (hsp)-60. The problem is that the structure of the hsp-60 protein strongly resembles that of other proteins located in other tissues of the body. In an individual with a defective immune system, cells of the immune system can mistakenly attack collagen type IV, thinking it is bacterial hsp-60. This is largely the hypothesized mechanism that leads to the development of rheumatoid arthritis and other autoimmune diseases. For example, there are other parts of the hsp-60 protein that resemble myelin basic protein. An inappropriate attack of this protein may lead to a process of demyelinization and multiple sclerosis. Yet another part of hsp-60 resembles dopamine decarboxylase. In some patients, an attack on this enzyme may lead to type I diabetes.We know now that many patients with glaucoma have an elevated hsp-60 titer. There is a good chance that the reason they have glaucoma is because there is something in the retina that could either be hsp 60 or a retinal protein that has some sequence homology with hsp-60, which the body mistakenly 'attacks' in its effort to rid itself of the offending pathogen with hsp60 on its surface. This is very compelling evidence that molecular mimicry plays a role in glaucoma.
- IgA and IgG immunoglobulin deposition in the ganglion cell layer
observed in the post-mortem examination of the eyes of a patient with
NPG and paraproteinemia.
In the 1980s, it was suspected that monoclonal paraproteins were the cause of several neuropathies. Therefore, neurologists performed biopsy studies on individuals with peripheral neuropathy and found that the neurons did indeed contain bound monoclonal antibodies. Glaucoma specialists obviously cannot biopsy optic nerves in living patients, but we did perform a postmortem study on the eyes of a patient with monoclonal gammopathy. Surprisingly, we found that monoclonal paraproteins were found in retinal ganglion cells as well as in other layers of the retina. This is another compelling clue that monoclonal gammopathies may be causative of glaucomatous neurogeneration, since the antibodies are found at a site where they can cause neuropathy. - abnormal T-cell findings in patients with glaucoma are similar
to those in patients with other autoimmune diseases.
Another important clue to showing that molecular mimicry seems to be a valid mechanism in patients with glaucoma is the antigens found on T cells in glaucoma patients. We found an increased frequency of certain antigens in T cells, particularly HLA and CD8, of normal-pressure glaucoma patients. These antigens are also upregulated in the T cells of patients with other autoimmune diseases (type 1 diabetes and rheumatoid arthritis). CD8+ lymphocytes are primarily involved in the immune defense to pathogens via their recognition of the major histocompatibility complex class I molecules of affected cells. The implication is that, whatever mechanism is involved by having certain T cells active in autoimmune diseases, patients with NPG appear to share this common finding with other common autoimmune diseases.
Despite the strength of these immunological associations, it is important to bear in mind that there is no evidence to confirm that retinal ganglion cell loss occurs as a direct result of aberrant humoral or cellular immunity in glaucoma patients. We are presently developing a model of experimental autoimmune glaucoma in order to determine whether immunization with cytotoxic autoantibodies will cause retinal cytotoxicity. The results thus far suggest that this is the case.
Unfortunately, little can be done therapeutically for patients who may
have presumed autoimmune glaucoma, apart from the traditional therapy of
trying to minimize the pressure-dependent component that may be injuring
RGCs. However, we must bear in mind that the treatment of autoimmune disease
is in its infancy, and will likely progress dramatically as the field of
peptidomimetics advances. In theory, we will eventually be able to identify
harmful autoantigens and neutralize them with molecules before they can
cause any damage by participating in adverse immune pathology.
If this body of work has demonstrated anything useful, it is that we must
pay attention to other mechanisms for potentially treating glaucomatous
neuropathy apart from pressure lowering. Autoimmunity is just one stress
condition that can destroy neurons.
References and Selected Reading
- Cartwright MJ, Grajewski AL, Friedberg ML, Anderson DR, Richards DW. 1992. Immune-related disease and normal-tension glaucoma: a case-control study. Arch Ophthalmol. 1992;110:500-502.
- Maruyama I, Ohguro H, Ikeda Y. 2000. Retinal ganglion cells recognized by serum autoantibody against gamma- enolase found in glaucoma patients. Invest Ophthalmol Vis Sci. 41:1657-1665.
- Romano C, Barrett DA, Li Z, Pestronk A, Wax MB. 1995. Anti-rhodopsin antibodies in sera from patients with normal-pressure glaucoma. Invest Ophthalmol Vis Sci. 36:1968-1975.
- Romano C, Li Z, Arendt A, Hargrave PA, Wax MB. 1999. Epitope mapping of anti-rhodopsin antibodies from patients with normal pressure glaucoma. Invest Ophthalmol Vis Sci. 40:1275-1280.
- Schwartz M, Kipnis J. 2001. Protective autoimmunity: regulation and prospects for vaccination after brain and spinal cord injuries. Trends Mol Med. 7:252-258.
- Tezel G, Edward DP, Wax MB. 1999. Serum autoantibodies to optic nerve head glycosaminoglycans in patients with glaucoma. Arch Ophthalmol. 117:917-924.
- Tezel G, Hernandez MR, Wax MB. 2000. Immunostaining of heat shock proteins in the retina and optic nerve head of normal and glaucomatous eyes. Arch Ophthalmol. 118:511-518.
- Tezel G, Seigel GM, Wax MB. 1998. Autoantibodies to small heat shock proteins in glaucoma. Invest Ophthalmol Vis Sci. 39:2277-2287.
- Tezel G, Wax MB. 2000. The mechanisms of hsp27 antibody-mediated apoptosis in retinal neuronal cells. J Neurosci. 20:3552-3562.
- Wax MB, Barrett DA, Pestronk A. 1994. Increased incidence of paraproteinemia and autoantibodies in patients with normal-pressure glaucoma. Am J Ophthalmol, 117:561-568.
- Wax MB, Tezel G, Edward PD. 1998. Clinical and ocular histopathological findings in a patient with normal-pressure glaucoma. Arch Ophthalmol. 116:993-1001.
- Wax MB, Tezel G, Kawase K, Kitazawa Y. 2001. Serum autoantibodies to heat shock proteins in glaucoma patients from Japan and the United States. Ophthalmology. 108(2):296-302.
- Wax MB, Tezel G, Saito I et al. 1998. Anti-Ro/SS-A positivity and heat shock protein antibodies in patients with normal-pressure glaucoma. Am J Ophthalmol. 125:145-157.
- Yang J, Patil RV, Yu H, Gordon M, Wax MB. 2001. T cell subsets and sIL-2R/IL-2 levels in patients with glaucoma. Am J Ophthalmol. 131:421-426.
- Yang J, Tezel G, Patil RV, Romano C, Wax MB. 2001. Serum autoantibody against glutathione S-transferase in patients with glaucoma. Invest Ophthalmol Vis Sci. 42:1273-1276.
- Yang J, Yang P, Tezel G, Patil RV, Hernandez MR, Wax MB. 2001. Induction of HLA-DR expression in human lamina cribrosa astrocytes by cytokines and simulated ischemia. Invest Ophthalmol Vis Sci. 42:365-371.
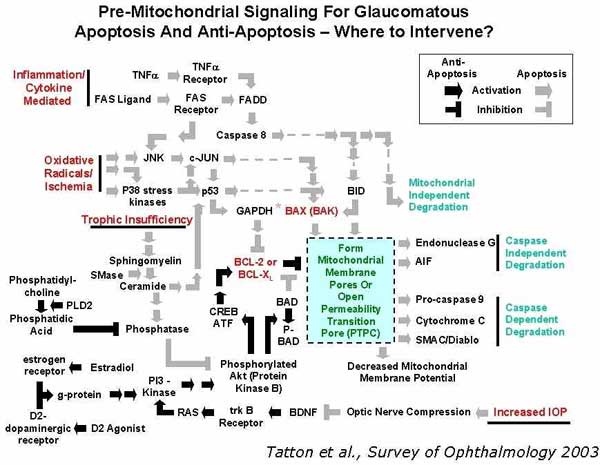 Figure 2
Figure 2